
NGS-perusteet blogikirjoitussarjan kolmannessa osassa selvitellään auki NGS-applikaatioissa käytettyjä ja etenkin sekvensointiajoon liittyviä parametrejä ja termejä. Ensimmäinen osa keskittyy käytännön labratyön esittelyyn, toinen osa NGS-applikaatioihin ja neljännessä osassa selvitän mitä tarkoittavat UDI:t (Unique Dual indices) ja UMI:t (molecular tags/barcodes) ja miten ne eroavat toisistaan.
Sekvensoinnin tunnuslukuja/terminologiaa
Sekvenssifragmentti (Read): sekvensoitu emäspätkä, välillä 50-250bp (yleisemmin käytössä olevien short read sekvensaattoreiden tuottama pituus). Tietyillä laitteistoilla pystytään sekvensoimaan jopa kymmenien tuhansien emästen pituisia jaksoja.
Read length – Sekvenssipätkän pituus, joka riippuu sekvensoinnissa käytettävien syklien määrästä. Yksi sykli lisää aina yhden emäksen sekvensoitavaan fragmenttiin. Esimerkiksi 100 bp fragmentin aikaansaamiseksi on käytetty 100 sykliä. Pidemmät sekvenssipätkien pituudet mahdollistavat tietenkin tarkemman sekvenssi-informaation, mutta tulevat tietenkin myös kalliimmaksi. 50 sykliä riittää yleensä esim. yleiseen RNA-profilointiin ja yli 100 bp eli kokonaisuudessaan laajempi output tarvitaan tarkempaan koko transkriptomin tai genomin analysointiin.
Aimed numbers of reads: Eri sekvensaattorit tuottavat eri määrän sekvenssejä; eri laitteistoilla on erilaiset kapasiteetit kuinka paljon dataa ja missä ajassa pystytään ajamaan. Mitä enemmän geenejä tai mitä syvemmin sekvensoidaan, sitä vähemmän näytteitä/per ajo. Yleisesti, isompien genomien ekspressioanalyysiin tarvitaan 25-30 miljoonaa readia kun taas esim. harvinaisempien transkriptien tai RNA-isoformien tunnistamiseen tarvitaan noin 100-200 miljoonaa readia jotta saadaan luotettava tulos.

Yksilötunniste (Barcode/Index): jokaisen näytteen identifioiva ”viivakoodi” = lyhyt sekvenssipätkä, joka lisätään kirjastontekovaiheessa. Eri barcode-kirjastot voidaan sekoittaa eli poolata yhteen ja näin mahdollistaa kustannustehokas sekvensointi & data-analyysi.
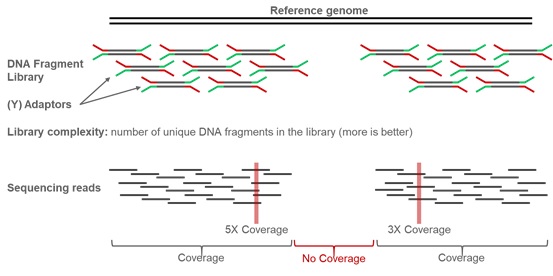
Kirjaston monimuotoisuus (Library complexity): Kirjaston erilaisten/uniikkien DNA fragmenttien määrä – Mitä enemmän, sitä parempi.
Mapped reads: Sekvenssipätkät jotka on pystytty onnistuneesti rinnastamaan referenssigenomiin (BAM-tiedostomuodossa).
Kattavuus tai lukusyvyys (Coverage): Referenssigenomiin rinnastettujen readien suhteellinen osuus eli kuinka monta kertaa tietty emäs on sekvensoitu (readit/sekvenssifragmentit eivät jakaudu tasaisesti koko genomin kattavasti). Esim. 80%, 0.8X, tarkoittaa että jokainen emäs on sekvensoitu keskimäärin 80 kertaa. Mitä parempi/korkeampi tämä luku on, sitä luotettavampi ja laadukkaampi sekvenssidata on. RNA:n kohdalla tämä on hankalampaa, koska eri geenien ekspressiot vaihtelevat; total number of mapped reads on siten parempi mittari.
Sekvenssiajon tyyppi, Single Read (SR) tai Paired End (PE):
Single Read SR:ssä sekvensoitava fragmentti luetaan/sekvensoidaan päästä päähän yhden kerran. Halvempi, nopeampi, ja yleensä käytössä profiloinnissa (esim. RNAseq ekspressioanalyysit, ChIP-Seq).
Paired End runs: Fragmentit sekvensoidaan molempiin suuntiin, mikä mahdollistaa tarkemman sekvenssi-informaation saamisen. Parempi esim. genomin uudelleenjärjestäytymisen (deleetiot, insertiot, inversiot) analysoinnissa sekä de novo sekvensoinnissa. Myös RNA-splice varianttien, metylaation ja SNP (small nucleotide polymorphism) -muutosten identifioinnissa on parempi käyttää paired-end ajoa (esim. 2×100 tai 2×150 read lengths).
Lisää hyödyllistä tietoa sekvensointiin liittyen löydät mm. tästä linkistä ja erilaisia NGS-ratkaisuja täältä.
Tutustu myös NGS-perusteet blogikirjoitussarjan muihin kirjoituksiin.
Blogin kirjoitti Sanna Siltanen, PhD.