
Tämän blogikirjoitussarjan tarkoituksena on kertoa perusteita NGS-sekvensoinnista, ja helpottaa erilaisten applikaatioiden valintaa. Ensimmäinen osa keskittyy käytännön labratyön esittelyyn ja toinen osa NGS-applikaatiohin. Kolmannessa osassa paneudumme sekvensoinnin terminologiaan ja neljännessä osassa selvitän mitä tarkoittavat UDI:t (Unique Dual indices) ja UMI:t (molecular tags/barcodes) ja miten ne eroavat toisistaan.
NGS workflow -Mitä ottaa huomioon?
Next generation sequencing (NGS) on jo pitkään käytössä ollut menetelmä perimän (DNA:n tai RNA:n) emäsjärjestyksen analysointiin. Sekvensoinnin avulla tutkitaan genomin virheitä ja variaatiota sekä geenien ilmentymisen (ekspression) muutoksia. NGS-sekvensointi perustuu massiiviseen rinnakkaissekvensointiin, jossa miljoonien sekvenssipätkien analysointi mahdollistuu kerralla yhdessä ajossa (parallel). Täten pystytään tutkimaan lukuisia geenejä ja kohteita useista eri näytteistä samanaikaisesti, mikä aiempaan Sanger-sekvensointiin tai esim. PCR-tekniikoihin verrattuna nopeuttaa potilasdiagnostiikkaa, uusien lajien tunnistusta jne., sekä on edullisempaa.
NGS:ää hyödynnetään sekä tutkimuksessa, että diagnostiikassa. NGS menetelmiä on käytössä esimerkiksi syöpädiagnostiikassa, patologian laboratorioissa ja perinnöllisten sairauksien tunnistukseen erikoistuneissa genetiikan laboratorioissa. Muita sovellusalueita ovat esimerkiksi farmakogenomiikka (lääkeaineiden vaikutukset/syöpähoito), metagenomiikka (mikrobiomiprojektit), mikrobiologia, virologia, epidemiologia (mikrobien; bakteerien, virusten tunnistus, antibioottiresistenssi, outbreak-epidemiat) ja de novo sekvensointi (uudet, tuntemattomat genomit & transkriptomit) ja genominlaajuiset CRISPR seulonnat.
Mutta mitä laboratoriossa sitten käytännössä tehdään?
Mitä tapahtuu näytteenoton ja lopulta pipelinesta ulos tulevan datan välillä, ja mitä tässä prosessissa tulee ottaa huomioon? Sekvensointiprotokollan valitsemiseen vaikuttaa olennaisesti millainen applikaatio ja näyte on kyseessä ja mitä etsitään/halutaan löytää. Onnistuneen tuloksen aikaansaamiseksi näytteenkäsittelyprosessi (Sample preparation) sisältää seuraavat vaiheet:
1. DNA:n tai RNA:n eristys & puhdistus
Tähän on olemassa useita erilaisia kaupallisia kittejä tai omia laboratorioprotokollia. Näytemateriaalina voi olla esim. solut, tuorekudos, veri, FFPE (formalin fixed paraffin embedded) kudospalat, plasma, soluvapaa DNA (cell free DNA), uloste sekä maaperä- tai vesinäyte. Tarkoituksena on saada mahdollisimman puhdas & edustava näyte aikaiseksi. RNA:n ollessa kyseessä se käännetään ensin cDNA:ksi, ja jos kiinnostuksen kohteena on esim. pienet RNA:t, ne on mahdollista valikoida jo tässä vaiheessa spesifisillä eristysmenetelmillä. Eristysmenetelmät voivat olla kolumni- tai liuospohjaisia. Jos näytemateriaali on hyvin vähäinen, esim. yksisolususpensiot (single cell samples) tai detekoidaan esim. viruksia pienistä hyönteisistä, DNA:ta/RNA:ta voi olla tarpeen monistaa (joko PCR:llä tai esim. isothermaalisella amplifikaatiolla).
2. DNA:n tai RNA:n laadun ja määrän mittaus
Puhdistetun DNA/RNA:n laatu ja määrä validoidaan spektrofotometrisilla tai fluoresenssiin perustuvilla menetelmillä (Qubit, Bioanalyzer). RNA:n ollessa kyseessä katsotaan RIN-arvoa, jolle voi olla vaatimuksena esim. arvo 8 hyvän sekvensointituloksen aikaansaamiseksi.
3. Näytteen pilkkominen
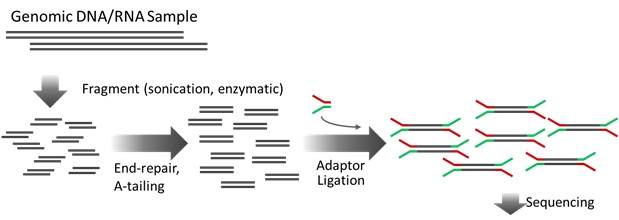
Tarvittaessa, riippuen käytetystä kirjastokitistä näyte fragmentoidaan, joko entsymaattisesti tai mekaanisesti sonikoinnin (shearing) avulla. Entsymaattiset menetelmät on nopeampia & helpompikäyttöisiä ja sonikoinnin avulla saadaan monimuotoisempi sekoitus erikokoisia fragmentteja. Jos näyte on jo valmiiksi hajonnutta (esim. FFPE-näytteet), fragmentointia ei välttämättä tarvita.
4. Kirjastojen valmistus:
DNA-kirjastot. Kirjastoprotokollien tarkoituksena on muodostaa sekvensoitavien fragmenttien pooli erilaisten entsymaattisten reaktioiden avulla. DNA-seqissä yleinen worklow sisältää kaksijuosteisen DNA:n päiden tasauksen (end-repair), mahdollisen adenylaation ja sekvenssiadaptoreiden liittämisen fragmentin molempiin päihin (adaptor ligation, nämä riippuvat käytetystä sekvensointilaitteistosta). Sekvensointiadaptorit kiinnittävät sekvensoitavan fragmentin sekvensointilaitteen flow celliin, sisältävät monistukseen tarvittavat primerit ja mahdollisesti myös näyte/molekyylitunnisteet eli barcodet.
RNA-kirjastot. RNA-applikaatioissa tehdään mahdollinen lähetti-RNA:n mRNA:n rikastus tai ribosomaalisen RNA:n depleetio totaali-RNA:sta, RNA:n fragmentaatio, käänteinen transkriptio (reverse transcription) , 1. juosteen synteesi (1st strand synthesis), 2. juosteen synteesi (second strand synthesis), adaptorien ligaatio & monistus (amplification). Ribosomaalisen RNA:n (rRNA) lisäksi on mahdollista depletoida eli poistaa muitakin/mitä tahansa häiritseviä transkriptejä. Kun se tehdään kirjastontekovaiheessa, säästetään näytettä ja myöhemmin sekvensointikustannuksia.
Kirjastojen puhdistus ja kvantitointi. Ennen sekvensointia DNA/RNA-kirjastot voi puhdistaa (size selection) ylimääräisistä reagensseista ja vääränkokoisista fragmenteista joita ei tarvitse sekvensoida, esim. primeri dimeerit. Tämä voidaan tehdä esim. geeli-elektroforeesin tai useimmiten magneetti-bead-pohjaisten menetelmien avulla.
Lopuksi, kirjastojen määrä mitataan fluoresenssiin perustuvilla menetelmillä tai qPCR:llä, ja laatu esim. Agilent TapeStation tai Bioanalyzer laitteistoilla.
5. Sekvensointiajo
Sekvensointi-platformeja/laitteistoja on useita erilaisia, enimmäkseen käytössä ovat Illuminan, Ion Torrentin ja Pacific Biosciences:n ratkaisut. Seuraavassa blogikirjoituksessa lisää sekvensointiin liittyvistä parametreistä.
6. Data-analyysi
Sekvensoinnin raakadata toimitetaan yleensä FASTQ-tiedostomuodossa. Analysoinnissa sekvensoidut fragmentit rinnastetaan referenssisekvenssiin, jolloin siitä poikkeavat emäkset voidaan tunnistaa. Käytettävät data-analyysityökalut tai pipelinet riippuvat tietenkin applikaatiosta, ja niitä on paljon erilaisia. Saatavilla on sekä ilmaisia käyttöliittymiä itsenäiseen datan analysointiin, valmiita, joidenkin kirjastokittien mukana tulevia protokollia/ohjelmia, että täysin asiakkaan tarpeiden mukaan räätälöityjä ratkaisuja.
7. Muita huomioonotettavia asioita
Erilaiset kontrollit: Sekvensoinnissa variaatiota aiheutuu enimmäkseen joko kirjastojen tekovaiheessa & monistamisen aikana tai sekvensointilaitteista johtuvista syistä, esim. huono sekvensointidatan laatu (poor base calling, bad sequencing cycles). Siksi onkin suositeltavaa käyttää useita näytereplikaatteja & ottaa mukaan erilaisia kontrolleja.
Esim. Acrometrixin Oncology HotSpot kontrolloi helposti muutoksia jokapäiväisessä työskentelyssä, lottien välillä ja laitteiden toiminnoissa. Tai RNA-töissä, on hyvä ottaa mukaan Spike-In RNA Variants (SIRVs). Targetoituja geenipaneeleja käytettäessä kontrolliksi & näytteiden jäljitykseen & identifiointiin on saatavilla oma paneelinsa.
Lisää hyödyllistä tietoa sekvensointiin liittyen löytyy ”Beginner’s Handbook to Next Generation Sequencing” ja erilaisia NGS-ratkaisuja meiltä!
Tutustu myös NGS-perusteet blogikirjoitussarjan muihin kirjoituksiin.
Blogin kirjoitti Sanna Siltanen, PhD.